
Gael, das Baby, das eines der teuersten Medikamente der Welt braucht und es immer noch nicht bekommen kann.

In Soacha, südlich von Bogotá, lebt Gael, ein gerade über ein Jahr altes Baby, dessen Leben sich zwischen Therapien, medizinischen Transporten und dem qualvollen Warten auf ein Medikament abspielt, das alles verändern könnte.
Gael leidet an spinaler Muskelatrophie (SMA), einer seltenen, degenerativen und potenziell tödlichen genetischen Erkrankung, die es ihm derzeit ermöglicht, nachts mit Hilfe eines BiPAP-Geräts (Bilevel Positive Airway Pressure) zu atmen und alle vier Monate unter Vollnarkose eine sehr teure Behandlung zu erhalten: Spinraza.
Ihre Eltern kämpfen jedoch um etwas anderes: den Zugang zur Gentherapie Zolgensma, die als das teuerste Medikament der Welt gilt und deren Kosten für eine einzige Anwendung über zwei Millionen Dollar betragen. Sie glauben, dass dies die einzige echte Chance für Gael ist, eines Tages frei zu gehen.
Edwin Puentes und Mónica Posada, Grafikdesigner und Eltern von Gael, hatten bereits einen neunjährigen Sohn, als sie beschlossen, ihre Familie zu vergrößern. Sie sagten, sie hätten es getan, um ihrem ältesten Sohn Martin einen Bruder zu schenken.

Auf dem Foto ist Gael mit seinem Bruder Martin und seinen Eltern Monica und Edwin zu sehen. Foto: Mit freundlicher Genehmigung von Gaels Eltern.
Monicas Schwangerschaft verlief reibungslos, ebenso wie die Kaiserschnittgeburt. Gael kam ohne Komplikationen auf die Welt. Doch nach etwa fünf Monaten begannen sie sich Sorgen zu machen: Ihr Baby konnte seinen Kopf nicht mehr halten, sich nicht umdrehen und nicht aufrecht sitzen. „Die Meilensteine der Entwicklung waren nicht vorhanden“, erinnert sich Edwin.
Ein Kinderarzt stellte Hypotonie fest, ein Symptom, das auf Muskelschwäche hinweist, und ordnete mehrere Tests an. Sie kamen alle normal heraus.
„Wir haben Ultraschalluntersuchungen, MRTs und Bluttests gemacht. Nichts davon konnte erklären, was wir sahen“, sagen sie. Schließlich wurden sie an die Genetik überwiesen. Dort wurden sie von Dr. Sandra Janeth Ospina Lagos, einer Genetikerin und Professorin an der National University, empfangen.
Die Diagnose Dr. Ospina vermutete sofort, dass es sich möglicherweise um eine schwere neuromuskuläre Erkrankung handelte. „ Gael war hypoton, konnte seinen Kopf nicht hochhalten, sein Gesicht wies die typischen myopathischen Züge auf, seine Reflexe waren praktisch nicht vorhanden und er nahm die sogenannte ‚Open-Book‘-Position ein“, erklärt er.
Er ordnete einen Gentest an, der seine Vermutung bestätigte: Gael litt an spinaler Muskelatrophie Typ 1B. SMA wird durch eine Mutation im SMN1-Gen verursacht, das für die Produktion des Survival-Motorneuron-Proteins (SMN) verantwortlich ist.
Ohne dieses Protein sterben Motoneuronen ab, was die Muskelbewegung, einschließlich Atmung und Schlucken, behindert . „Motorneuronen und Muskelzellen regenerieren sich nicht. Gehen sie verloren, gibt es kein Zurück mehr“, erklärt der Genetiker.
Die Krankheit ist erblich und autosomal-rezessiv, das heißt, beide Elternteile müssen Träger des mutierten Gens sein, damit ihr Kind daran erkrankt. Bei keinem von ihnen waren sichtbare Erscheinungen zu sehen.
„Es bestand eine 25-prozentige Wahrscheinlichkeit, ein Kind mit der Krankheit zu bekommen, und ohne einen speziellen Gentest konnte man das im Voraus nicht wissen.“
Als die Diagnose bestätigt wurde, war Gael zehn Monate alt. Zu diesem Zeitpunkt war er bereits auf die Atmung angewiesen, eine dauerhafte Beatmung war jedoch noch nicht erforderlich.
Dr. Ospina aktivierte alle Protokolle und begann mit der Anberaumung einer Sitzung des Ärzteausschusses. „Die Krankheit schreitet sehr schnell voran. Je früher die Behandlung beginnt, desto besser sind die Ergebnisse“, sagt er.
Der erste Schritt bestand darin, Gael in ein Krankenhaus einzuweisen, wodurch die Genehmigungen der EPS für die Behandlung mit Spinraza, einem Medikament, das direkt auf das Rückenmark aufgetragen wird, beschleunigt wurden .
Die Maßnahmen des Arztes zeigten Wirkung: Innerhalb weniger Wochen erhielt Gael seine erste Injektion. Heute hat er drei.
Ein schmerzhafter Eingriff Jede Anwendung von Spinraza ist ein Mini-Eingriff. Gael muss vollständig betäubt, in die fötale Position gebracht und mit einer Nadel in den Rückenmarksraum gelangt werden, und es muss Gehirn-Rückenmarks-Flüssigkeit entnommen werden, bevor das Medikament injiziert wird.
Zu Beginn der Behandlung wird die Dosis alle 15 Tage verabreicht; Nach der dritten Dosis wird die Dosis einmal im Monat und dann lebenslang alle 4 Monate verabreicht.
Gael muss sich dann lebenslang alle vier Monate dem Eingriff unterziehen. „Wir stehen um drei Uhr morgens auf, fahren nach Bogotá und sehen ihm beim Einschlafen zu, ohne zu wissen, ob alles gut wird. In der Genehmigung, die wir unterschrieben haben, steht, dass viel passieren kann“, sagt Mónica.
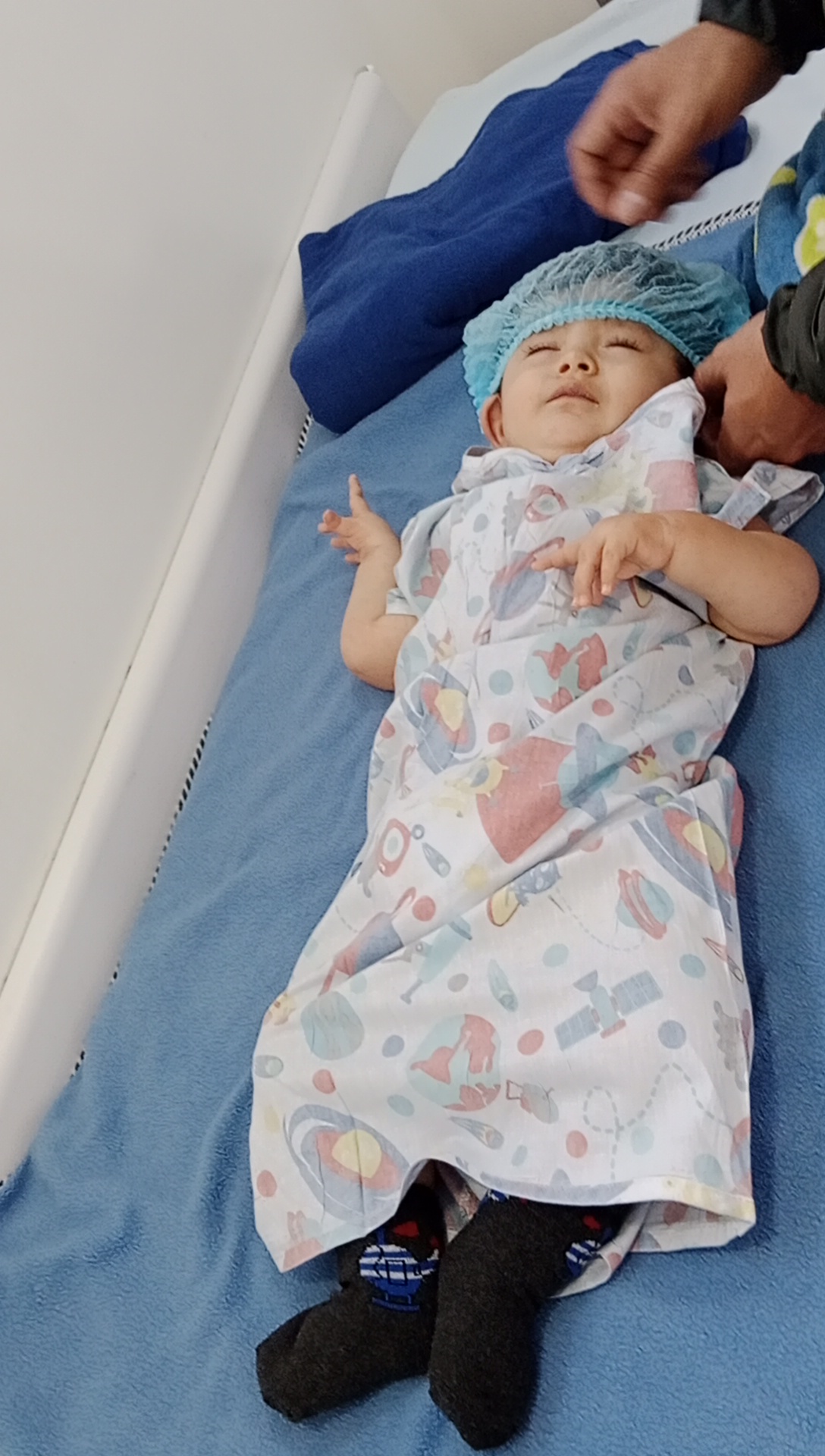
Gael bereitet sich auf die Verabreichung einer Spinraza-Dosis vor. Foto: Mit freundlicher Genehmigung von Gaels Eltern.
Trotz der Komplexität des Prozesses hat Gael die Behandlung gut vertragen. Es gab keine ernsthaften Nebenwirkungen und seine Eltern bemerkten leichte Verbesserungen, wie etwa eine stärkere Nackenmuskulatur und eine verbesserte Beweglichkeit. Sie wissen aber auch, dass diese Behandlung keine Heilung ist.
„Spinraza hilft, eine Verschlechterung so schnell zu verhindern, aber Gael ist immer noch gefährdet“, warnt Edwin. „Was Ihr Leben wirklich verändern könnte, ist Zolgensma.“
Hoffnung: Eine einzigartige Gentherapie Zolgensma ist eine revolutionäre Gentherapie, die direkt auf die Ursache der Krankheit einwirkt. Dabei wird über ein modifiziertes Virus eine funktionsfähige Kopie des SMN1-Gens in den Körper eingebracht, die dann das Nervensystem erreicht und das fehlende Protein produziert.
Im Gegensatz zu Spinraza wird es nur einmal verabreicht und kann den Krankheitsverlauf radikal verändern.
Allerdings gibt es dafür Bedingungen: Das Kind muss jünger als zwei Jahre sein, darf nicht dauerhaft beatmet werden und darf keinen Antikörper gegen das bei der Therapie verwendete Virus aufweisen. Gael erfüllt diese Kriterien.
„Er ist ein geeigneter Kandidat. Aber es gibt in Kolumbien keine Zentren, die diese Therapie anbieten, obwohl sie bereits vom Invima (Nationales Institut für Medizinische Forschung) zugelassen ist. Sie wurde im Land noch keinem einzigen Patienten verabreicht“, erklärt Dr. Ospina.
Zolgensma kostet rund 2,1 Millionen Dollar. Obwohl die Registrierung in Kolumbien genehmigt ist, wird sie bisher in keinem Krankenhaus verwendet.
„Wir wissen, dass es in Ländern wie Argentinien Kinder gibt, die diese Hilfe bereits erhalten haben und beeindruckende Fortschritte gemacht haben. Aber hier ist alles schwieriger“, beklagt Mónica. „Wir brauchen ein Gesundheitssystem, das den ersten Schritt wagt“, fügte er hinzu.
Mittlerweile hat die Familie eine Kampagne zur Beschaffung der Medikamente gestartet. „Manchmal werden wir in den sozialen Medien angegriffen. Sie sagen schreckliche Dinge. Aber wir wollen unserem Sohn einfach nur die Chance geben zu leben, zu laufen und zu spielen“, sagt Edwin.
Martín, Gaels älterer Bruder, hat angesichts dieser ganzen Situation eine Beschützerrolle übernommen. „Sie deckt ihn zu, spielt mit ihm, kitzelt ihn. Obwohl sie weiß, dass sie nicht an seine Seite rennen kann, begleitet sie ihn mit einer Liebe, die uns das Herz bricht.“
Therapien, Verfahren und Resistenzen Zusätzlich zur medizinischen Behandlung erhält Gael wöchentlich drei Arten von Therapien: Physiotherapie, Ergotherapie und Schlucktherapie. Seine Familie führt sie mit professioneller Unterstützung durch. Alles ist auf seine Entwicklung ausgerichtet.

Für Edwin und Monica ist es schwierig, ihre Zeit zwischen Therapien und medizinischen Verfahren aufteilen zu müssen. Foto: Mit freundlicher Genehmigung von Gaels Eltern.
„Die Grippe kann tödlich sein, deshalb benutzen wir keine öffentlichen Verkehrsmittel, tragen ständig Masken und fahren in Selbstbedienungs-Vans. Wir erhalten dafür keine finanzielle Unterstützung von der EPS“, erklären ihre Eltern.
Die Tage vergehen voller Formulare, Prüfungen, Petitionen, Papierkram und äußerster Sorgfalt. „Wir sind Grafikdesigner, aber Monica hat aufgehört zu arbeiten, um sich um Gael zu kümmern. Ich mache auch von zu Hause aus, was ich kann“, sagt Edwin.
Darüber hinaus müssen sie die Therapien für Martín koordinieren, bei dem eine Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS) diagnostiziert wurde.
Die Bedürfnisse zweier Kinder unter einen Hut zu bringen , ist eine enorme Belastung, aber Edwin und Monica versuchen jeden Tag, diese zu bewältigen.
In Kolumbien gibt es keine Exzellenzzentren für seltene Krankheiten wie SMA. Die Verabreichung von Behandlungen wie Zolgensma erfordert Infrastruktur, Wissen und Protokolle.
„Wir versuchen, die beste und sicherste Option zu wählen. Aber wir träumen auch davon, den entscheidenden Schritt in Richtung Heilung zu wagen“, sagt Dr. Ospina.
Sie weiß, dass Gaels Leben nicht gesichert ist. Ohne Behandlung ist die Lebenserwartung bei SMA Typ 1 sehr kurz. Babys sterben oft vor dem zweiten Lebensjahr aufgrund von Atemwegskomplikationen.
„Gael braucht jetzt nachts Unterstützung, was auf eine Beteiligung der Atemmuskulatur hindeutet. Aber es ist noch Zeit“, betont Ospina.
Gael ist ein Jahr und zwei Monate alt. Die Uhr tickt. Jeder Tag, der ohne die Einnahme von Zolgensma vergeht, ist ein Fenster, das sich schließt. Seine Eltern wissen es.
„ Wir wollen nicht, dass er sein Leben lang alle vier Monate operiert werden muss. Wir wollen, dass er ohne Angst atmen, rennen und singen kann. Er liebt Musik. Wir träumen davon, ihn tanzen, springen und frei sein zu sehen“, sagt Monica mit brechender Stimme.
Dies gilt nicht nur für Gael. Es ist die Geschichte von immer mehr Menschen mit seltenen Krankheiten in Kolumbien, die keinen Zugang zu Behandlungen haben, die zwar vorhanden sind, im Land aber noch nicht angewendet werden. Gael wartet immer noch. Und seine Eltern kämpfen weiter, damit er die beste Behandlung erhält.
Für weitere Informationen kontaktieren Sie bitte Edwin Puentes unter +57 321 3486405 oder über die sozialen Medien @todoscongaelpuentes.
ANGELA MARÍA PÁEZ RODRÍGUEZ - SCHULE FÜR MULTIMEDIAJOURNALISMUS EL TIEMPO.
eltiempo




