
Gael, el bebé que necesita uno de los medicamentos más caros del mundo y aún no puede conseguirlo

En Soacha, al sur de Bogotá, vive Gael, un bebé de poco más de un año cuya vida transcurre entre terapias, traslados médicos y la espera angustiante de un medicamento que podría cambiarlo todo.
Gael tiene atrofia muscular espinal (AME), una enfermedad genética rara, degenerativa y potencialmente mortal, que hoy lo obliga a respirar con ayuda de un BiPAP (Bilevel Positive Airway Pressure) por las noches y a recibir cada cuatro meses, bajo anestesia general, un tratamiento de altísimo costo: Spinraza.
Sin embargo, sus padres luchan por algo más: acceder a la terapia génica Zolgensma, considerada la medicina más cara del mundo, con un precio que supera los dos millones de dólares por única aplicación. Creen que es la única posibilidad real de que Gael camine algún día.
Edwin Puentes y Mónica Posada, diseñadores gráficos y padres de Gael, tenían ya un hijo de nueve años cuando decidieron agrandar la familia. Lo hicieron, dicen, para regalarle a su hijo mayor, Martín, un hermano.

En la foto Gael junto a su hermano Martin y sus padres Mónica y Edwin. Foto:Cortesía de los padres de Gael.
El embarazo de Mónica transcurrió sin sobresaltos, al igual que el nacimiento por cesárea. Gael llegó al mundo sin complicaciones. Pero hacia los cinco meses, algo empezó a inquietarlos: su bebé no sostenía la cabeza, no giraba, no se sentaba. “Los hitos del desarrollo no estaban”, recuerda Edwin.
Una pediatra detectó hipotonía, un síntoma que señala debilidad muscular, y le ordenó varios exámenes. Todos salieron normales.
“Hicimos ecografías, resonancias, exámenes de sangre. Nada explicaba lo que veíamos”, dicen. Finalmente, fueron remitidos a genética. Allí los recibió la doctora Sandra Janeth Ospina Lagos, médica genetista y profesora de la Universidad Nacional.
El diagnósticoLa doctora Ospina sospechó de inmediato la posibilidad de una enfermedad neuromuscular grave. “Gael era hipotónico, no sostenía la cabeza, su rostro tenía rasgos típicos de miopatía, sus reflejos estaban prácticamente ausentes y adoptaba la llamada posición de ‘libro abierto’”, explica.
Ordenó una prueba genética que confirmó su intuición: Gael tenía atrofia muscular espinal tipo 1B. La AME es causada por una mutación en el gen SMN1, responsable de producir la proteína de supervivencia de la motoneurona (SMN).
Sin esta proteína, las neuronas motoras mueren, lo que impide el movimiento muscular, incluyendo la respiración y la deglución. “Las neuronas motoras y las células musculares no se regeneran. Si se pierden, no hay vuelta atrás”, explica la genetista.
La enfermedad es hereditaria y autosómica recesiva, es decir, ambos padres deben portar el gen mutado para que el hijo la desarrolle. Ninguno de ellos tenía manifestaciones visibles.
“Había un 25 por ciento de probabilidad de tener un hijo con la enfermedad y no había forma de saberlo antes sin una prueba genética específica”.
Cuando se confirmó el diagnóstico, Gael tenía diez meses. Para entonces, ya necesitaba ayuda para respirar, aunque aún no ha requerido ventilación permanente.
La doctora Ospina activó todos los protocolos, iniciando por citar una junta médica. “La enfermedad avanza muy rápido. Entre más temprano se inicie el tratamiento, mejores son los resultados”, afirma.
El primer pasó fue internar a Gael en un hospital, esto permitió acelerar las autorizaciones de la EPS para iniciar con Spinraza, un medicamento que se aplica directamente en la médula espinal.
Las acciones de la doctora funcionaron: en cuestión de semanas, Gael recibió la primera de sus inyecciones. Hoy lleva tres.
Un procedimiento dolorosoCada aplicación de Spinraza es una mini cirugía. Hay que anestesiar por completo a Gael, colocarle en posición fetal, acceder con una aguja al espacio medular y extraer líquido cefalorraquídeo para luego inyectar el medicamento.
Al principio del tratamiento tiene una periodicidad de una dosis cada 15 días, después de la tercera dosis, viene 1 al mes y luego cada 4 meses de por vida.
Luego, Gael tendrá que someterse al procedimiento cada cuatro meses, de por vida. “Nos levantamos a las tres de la mañana, viajamos hasta Bogotá y lo vemos dormirse sin saber si todo saldrá bien. La autorización que firmamos dice que pueden pasar muchas cosas”, dice Mónica.
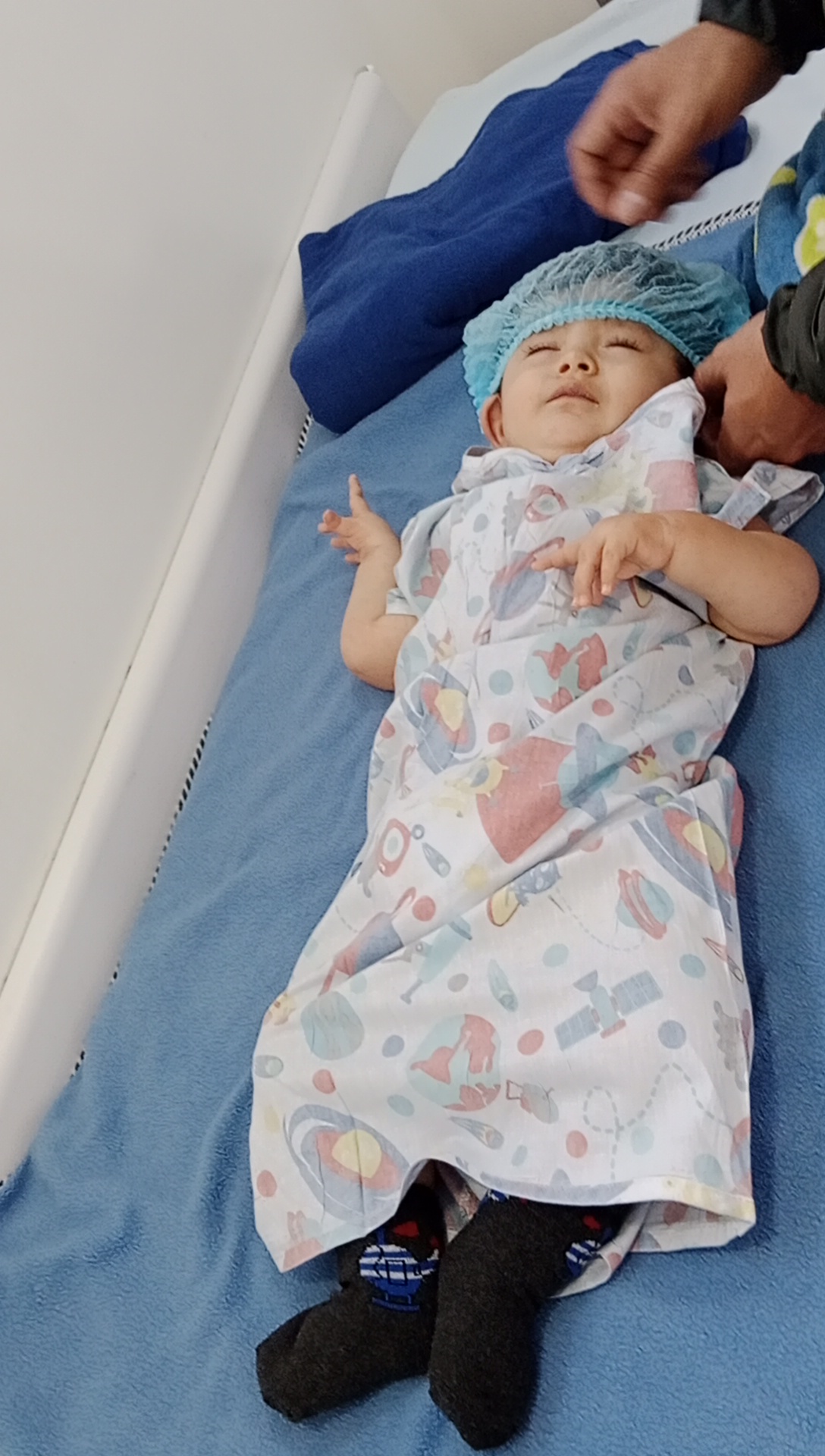
Gael en la preparación para recibir una dosis de Spinraza. Foto:Cortesía padres de Gael.
Pese a lo complejo del proceso, Gael ha tolerado bien el tratamiento. No ha presentado efectos secundarios graves, y sus padres notan pequeñas mejoras como por ejemplo: más fuerza en el cuello, más movimientos. Pero también saben que este tratamiento no es una cura.
“Spinraza ayuda a que no se deteriore tan rápido, pero Gael sigue en riesgo”, advierte Edwin. “Lo que realmente podría cambiar su vida es Zolgensma”.
La esperanza: una terapia génica única Zolgensma es una terapia génica revolucionaria que actúa directamente sobre la causa de la enfermedad. Introduce una copia funcional del gen SMN1 en el cuerpo a través de un virus modificado, que logra llegar al sistema nervioso y producir la proteína faltante.
A diferencia de Spinraza, se administra una sola vez y puede modificar radicalmente la evolución de la enfermedad.
Pero hay condiciones: el niño debe tener menos de dos años, no estar en soporte ventilatorio permanente y ser negativo para anticuerpos contra el virus usado en la terapia. Gael cumple esos criterios.
“Él es candidato. Pero en Colombia no hay centros que apliquen la terapia, aunque ya está aprobada por el Invima. Nunca se ha administrado a un solo paciente en el país”, explica la doctora Ospina.
Zolgensma cuesta alrededor de 2.1 millones de dólares. Aunque su registro está aprobado en Colombia, ningún hospital la aplica aún.
“Sabemos que en países como Argentina ya hay niños que la han recibido y han mostrado avances impresionantes. Pero acá todo es más difícil”, lamenta Mónica. “Necesitamos que el sistema de salud se atreva a dar el primer paso”, agrega.
Mientras tanto, la familia ha iniciado una campaña para conseguir el medicamento. “La gente en redes a veces nos ataca. Dicen cosas horribles. Pero nosotros solo queremos darle a nuestro hijo la oportunidad de vivir, de caminar, de jugar”, dice Edwin.
Martín, el hermano mayor de Gael, ha asumido un rol protector ante toda esta situación. “Lo arropa, juega con él, le hace cosquillas. Aunque sabe que no puede correr a su lado, lo acompaña con un amor que nos parte el alma”.
Terapias, trámites y resistenciaAdemás del tratamiento médico, Gael recibe tres tipos de terapias semanales: física, ocupacional y para la deglución. Su familia las realiza con apoyo profesional. Todo está centrado en su desarrollo.

Para Edwin y Mónica resulta complejo tener que repartir su tiempo entre terapias y tramites médicos. Foto:Cortesía padres de Gael.
“Una gripa puede ser fatal, por eso no salimos al transporte público, usamos tapabocas todo el tiempo, nos movemos en carros de aplicación. No recibimos apoyo económico de la EPS para esto”, aclaran sus padres.
Los días transcurren entre formularios, exámenes, derechos de petición, trámites y cuidados extremos. “Somos diseñadores gráficos, pero Mónica dejó de trabajar para cuidar a Gael. Yo también hago lo que puedo desde casa”, dice Edwin.
Además, tienen que coordinar las terapias de Martín, quien fue diagnosticado con trastorno por déficit de atención e hiperactividad (TDAH).
Poder equilibrar las necesidades de los dos niños resulta en una carga enorme, pero Edwin y Mónica lo intentan sortear día a día.
En Colombia, no existen centros de excelencia para enfermedades huérfanas como la AME. La administración de tratamientos como Zolgensma requiere infraestructura, conocimiento y protocolos.
“Tratamos de elegir la mejor opción disponible, la más segura. Pero también soñamos con poder dar ese salto hacia la cura”, dice la doctora Ospina.
Ella sabe que la vida de Gael no está garantizada. Sin tratamiento, la AME tipo 1 tiene una expectativa de vida muy corta. Los bebés suelen morir antes de los dos años por complicaciones respiratorias
“Gael ya necesita un soporte nocturno, lo cual indica compromiso de los músculos respiratorios. Pero está a tiempo”, insiste Ospina.
Gael tiene un año y dos meses. El reloj corre. Cada día que pasa sin recibir Zolgensma es una ventana que se cierra. Sus padres lo saben.
“No queremos que tenga que pasar por una cirugía cada cuatro meses toda la vida. Queremos que respire sin miedo, que corra, que cante. Le encanta la música. Soñamos con verlo bailar, saltar, ser libre”, dice Mónica, con la voz quebrada.
Este no es solo el caso de Gael. Es la historia de más personas con enfermedades raras en Colombia que no logran acceder a tratamientos que existen, pero aun no son aplicados en el país. Gael sigue esperando. Y sus padres siguen luchando para que él pueda recibir el mejor tratamiento.
Para mayor información, puede contactarse con Edwin Puentes al número celular +57 321 3486405 o por medio de las redes sociales @todoscongaelpuentes.
ÁNGELA MARÍA PÁEZ RODRÍGUEZ - ESCUELA DE PERIODISMO MULTIMEDIA EL TIEMPO.
eltiempo




